
I'm looking for good students with an interest in theoretical chemistry and/or (bio)inorganic chemistry, nanomaterials or prebiotic chemistry.
The fellowship is related to the "Correlating functionality (CorrFun)" project (PID2020-114548GB-I00, Sep. 2021-Sep. 2024) from the Ministerio de Ciencia e Innovación (MCI). The official recruitment will be done through the formal procedures at the Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR) , but I will be pre-screening candidates until November 14, 2022.
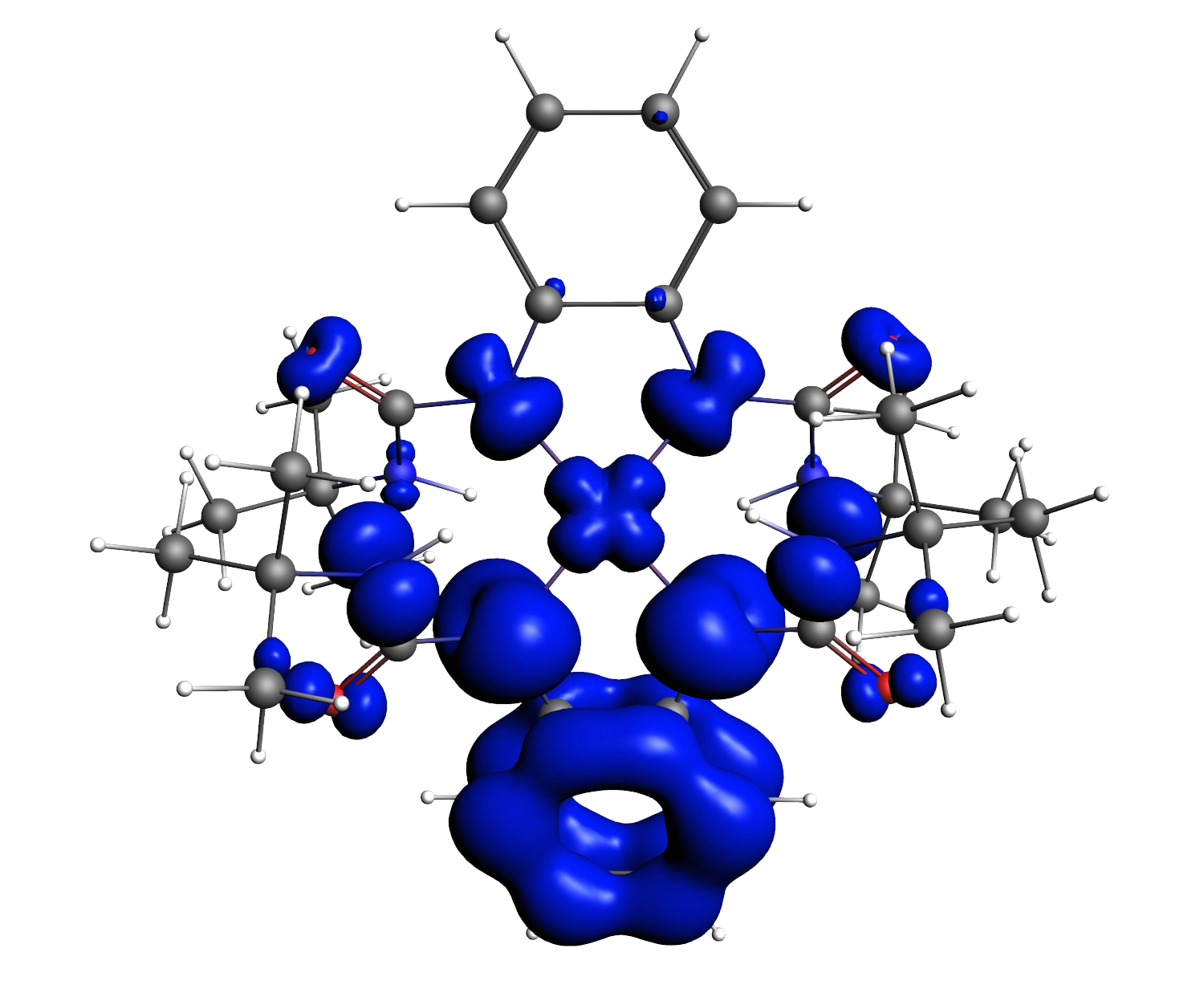
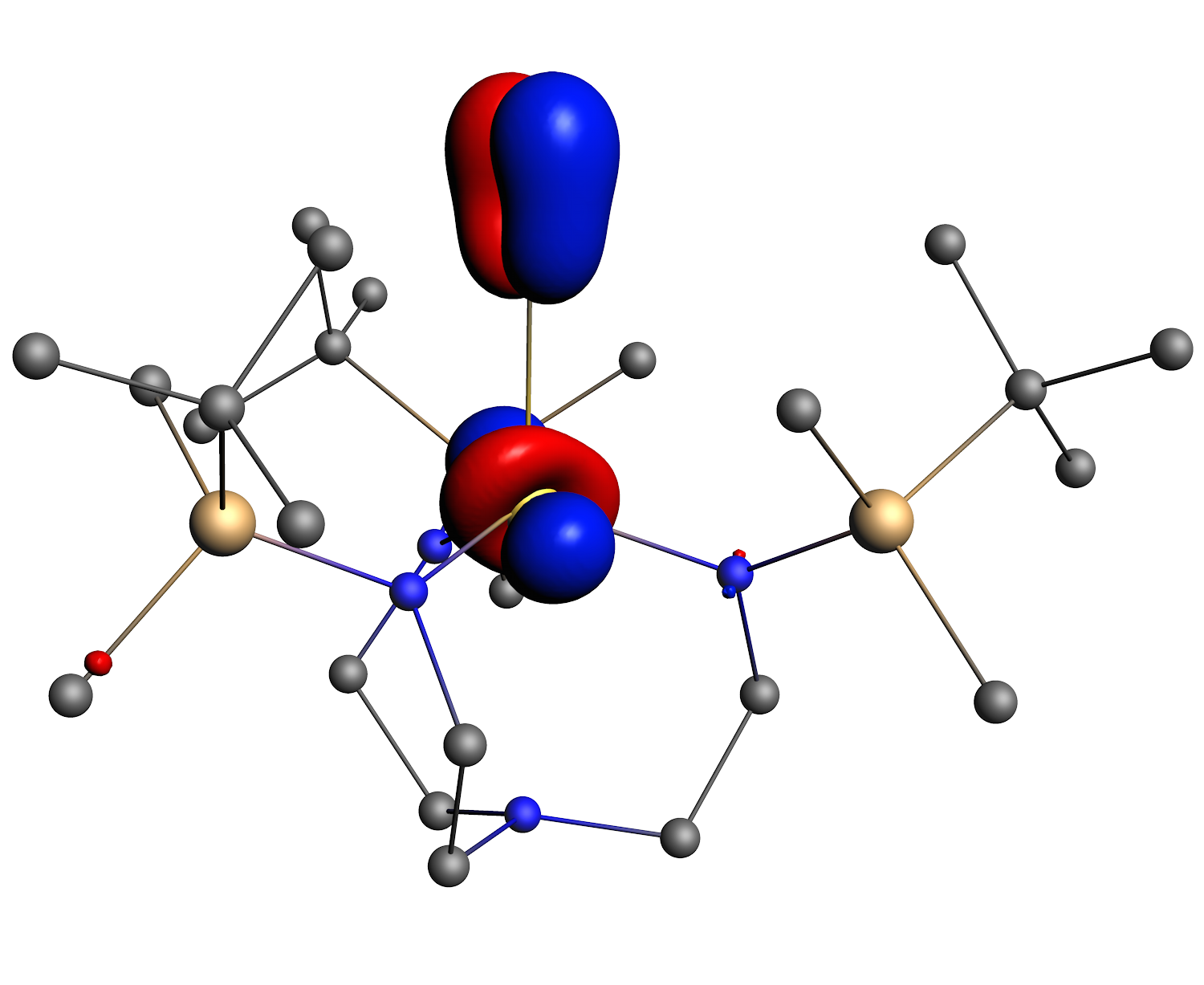
Chemistry can (or should) be defined as the discipline of transformation, where molecules meet, interact and then depart completely reshaped; these processes can be enhanced or made more selective through the implication of (transition) metals. The first-row transition metals (Sc-Cu) play a special role in this, in the sense that they are earth-abundant (allowing for sustainable, or green, catalysis), and in general show a larger sensitivity to how the electrons are distributed over the d-orbitals than the corresponding transition-metals in higher rows of the Periodic Table. Correlating experimental observations (that these transition-metals induce) with the molecular structure can be facilitated by computational chemistry, provided that sufficiently accurate methods are used. This is in particular true for spin and oxidation states of these transition-metals, for which density functionals have been developed in the Swart lab. Recently, a modification of the correlation part of the exchange-correlation energy was explored, which in preliminary results shows very promising progress towards an universal density functional. Within this project, these ideas for a new correlation functional will be further explored, and used as input to create a DFTB parameterization for first-row transition metals. These new computational tools will be applied to a range of systems in collaboration with experimental labs, to study the mechanisms of oxidation reactions, the effect of the ligand and/or oxidation state on them, and how new nanomaterials can be constructed. We will be carrying out computational chemistry experiments to characterize intermediates and transition structures, to determine the most favorable reaction path. An important role in these characterizations is played by computational spectroscopy (IR, Raman, EPR, Mössbauer, NMR, XAS), which allows for direct comparison with experimental spectra, and hence the confirmation (or negation) of the species involved in the reactions. This project is therefore providing new computational tools, and new chemical insights from which many research groups in the field will benefit.